library(gstudio)
data(arapat)5 Genetic Diversity
This chapter covers estimation of genetic diversity statistics using the genetic_diversity() wrapper and its underlying per-locus functions.
5.1 The genetic_diversity() Wrapper
The genetic_diversity() function is the main entry point for diversity estimation. It dispatches to specific estimators based on the mode parameter:
| Mode | Statistic | Description |
|---|---|---|
"A" |
Allelic richness | Number of alleles |
"Ae" |
Effective alleles | \(1 / \sum p_i^2\) |
"A95" |
A (5% threshold) | Alleles with frequency >= 0.05 |
"He" |
Expected heterozygosity | \(1 - \sum p_i^2\) |
"Ho" |
Observed heterozygosity | Proportion of heterozygotes |
"Hes" |
Corrected He | Nei’s small-sample corrected He |
"Hos" |
Corrected Ho | Nei’s small-sample corrected Ho |
"Fis" |
Inbreeding coefficient | \(1 - H_o/H_e\) |
"Pe" |
Polymorphic index | Whether the locus is polymorphic |
5.2 Global (Unstratified) Estimates
Without a stratum, diversity is estimated across all individuals:
genetic_diversity(arapat, mode = "A") Locus A
1 LTRS 2
2 WNT 5
3 EN 5
4 EF 2
5 ZMP 2
6 AML 13
7 ATPS 10
8 MP20 19genetic_diversity(arapat, mode = "Ae") Locus Ae
1 LTRS 1.995623
2 WNT 2.880450
3 EN 1.814656
4 EF 1.773533
5 ZMP 1.513877
6 AML 5.860583
7 ATPS 3.563347
8 MP20 5.511837genetic_diversity(arapat, mode = "He") Locus He
1 LTRS 0.4989034
2 WNT 0.6528320
3 EN 0.4489313
4 EF 0.4361538
5 ZMP 0.3394444
6 AML 0.8293685
7 ATPS 0.7193649
8 MP20 0.8185723genetic_diversity(arapat, mode = "Ho") Locus Ho
1 LTRS 0.23691460
2 WNT 0.27840909
3 EN 0.20555556
4 EF 0.14404432
5 ZMP 0.15454545
6 AML 0.37058824
7 ATPS 0.08539945
8 MP20 0.446927375.3 Per-Stratum Estimates
Provide a stratum argument to compute diversity within each population:
genetic_diversity(arapat, stratum = "Species", mode = "He") Stratum Locus He
2 Cape LTRS 0.14720000
3 Cape WNT 0.23374726
4 Cape EN 0.42000000
5 Cape EF 0.06444444
6 Cape ZMP 0.00000000
7 Cape AML 0.53312835
8 Cape ATPS 0.11546667
9 Cape MP20 0.36791111
10 Mainland LTRS 0.38850309
11 Mainland WNT 0.03277778
12 Mainland EN 0.50285714
13 Mainland EF 0.27119377
14 Mainland ZMP 0.09500000
15 Mainland AML 0.53854875
16 Mainland ATPS 0.25038580
17 Mainland MP20 0.80670340
18 Peninsula LTRS 0.42591805
19 Peninsula WNT 0.50613781
20 Peninsula EN 0.17229600
21 Peninsula EF 0.48979592
22 Peninsula ZMP 0.41492187
23 Peninsula AML 0.72842417
24 Peninsula ATPS 0.52255763
25 Peninsula MP20 0.69148800genetic_diversity(arapat, stratum = "Species", mode = "Ae") Stratum Locus Ae
2 Cape LTRS 1.172608
3 Cape WNT 1.305052
4 Cape EN 1.724138
5 Cape EF 1.068884
6 Cape ZMP 1.000000
7 Cape AML 2.141916
8 Cape ATPS 1.130540
9 Cape MP20 1.582056
10 Mainland LTRS 1.635331
11 Mainland WNT 1.033889
12 Mainland EN 2.011494
13 Mainland EF 1.372107
14 Mainland ZMP 1.104972
15 Mainland AML 2.167076
16 Mainland ATPS 1.334020
17 Mainland MP20 5.173397
18 Peninsula LTRS 1.741912
19 Peninsula WNT 2.024856
20 Peninsula EN 1.208161
21 Peninsula EF 1.960000
22 Peninsula ZMP 1.709173
23 Peninsula AML 3.682213
24 Peninsula ATPS 2.094494
25 Peninsula MP20 3.2413655.4 Individual Estimator Functions
Each diversity metric has a standalone function that operates on a single locus vector.
5.4.1 Allelic Richness
A(arapat$LTRS)A
2 5.4.2 Effective Number of Alleles
Ae(arapat$LTRS) Ae
1.995623 5.4.3 Expected Heterozygosity
He(arapat$LTRS) He
0.4989034 5.4.4 Observed Heterozygosity
Ho(arapat$LTRS) Ho
0.2369146 5.4.5 Inbreeding Coefficient
Fis(arapat$LTRS) Fis
0.5251293 5.4.6 Exclusion Probability
Pe(arapat$LTRS)[1] 0.49890345.5 Corrected Heterozygosity (Nei’s)
The Hes and Hos modes require a stratum because they apply a small-sample correction per subpopulation:
genetic_diversity(arapat, stratum = "Species", mode = "Hes") Locus Hes
1 LTRS 0.3233431
2 WNT 0.2606681
3 EN 0.3687470
4 EF 0.2781409
5 ZMP 0.1731159
6 AML 0.6108282
7 ATPS 0.2999070
8 MP20 0.6284672
9 Multilocus 0.36790225.6 Multilocus Diversity
The multilocus_diversity() function computes a summary across all loci:
multilocus_diversity(arapat)[1] 0.83746565.7 Selecting Specific Loci
You can restrict the analysis to a subset of loci:
genetic_diversity(arapat, loci = c("LTRS", "WNT"), mode = "He") Locus He
1 LTRS 0.4989034
2 WNT 0.65283205.8 Comparing Populations
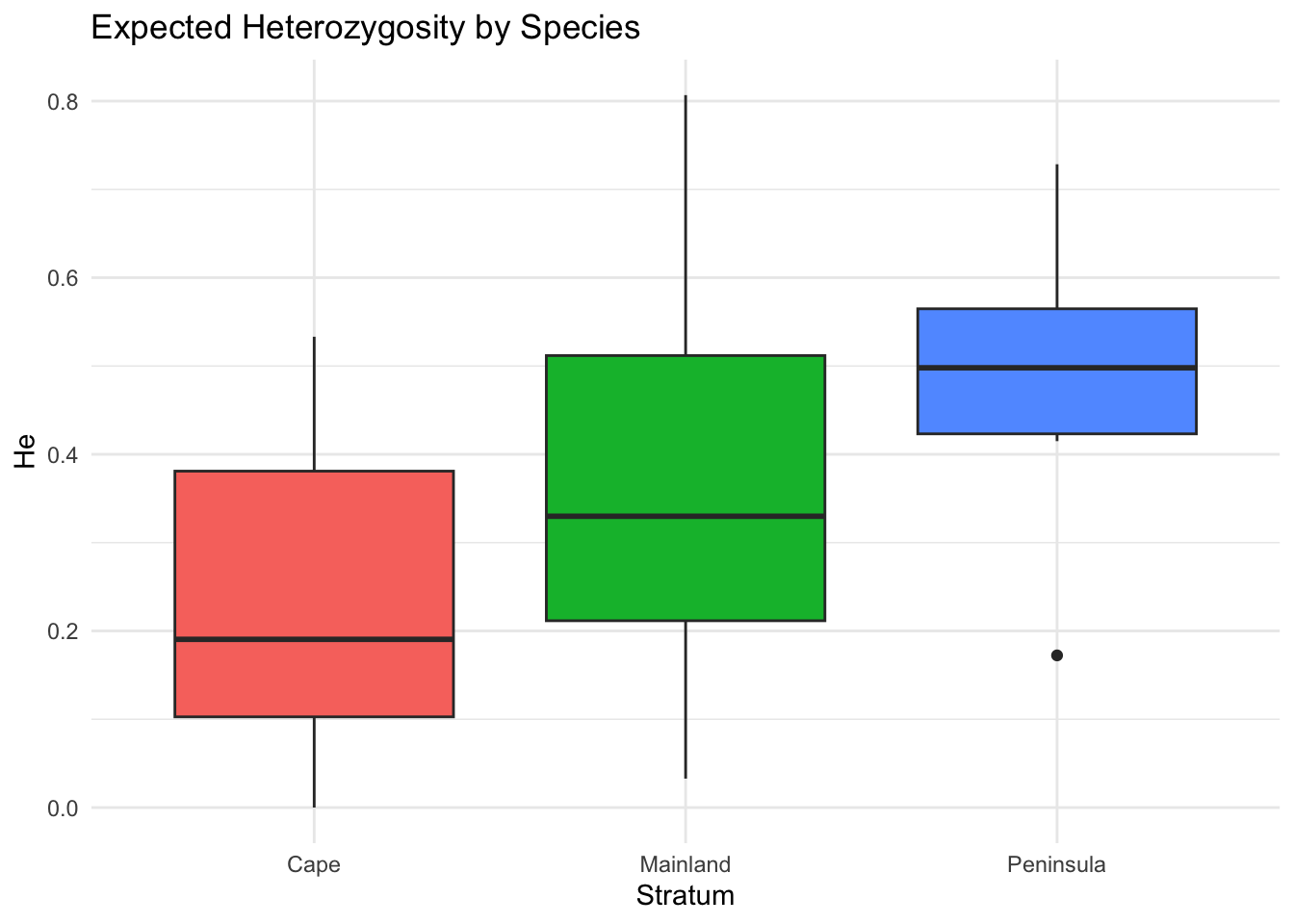
A common workflow is to estimate diversity per population, then compare:
he_by_pop <- genetic_diversity(arapat, stratum = "Species", mode = "He")
he_by_pop Stratum Locus He
2 Cape LTRS 0.14720000
3 Cape WNT 0.23374726
4 Cape EN 0.42000000
5 Cape EF 0.06444444
6 Cape ZMP 0.00000000
7 Cape AML 0.53312835
8 Cape ATPS 0.11546667
9 Cape MP20 0.36791111
10 Mainland LTRS 0.38850309
11 Mainland WNT 0.03277778
12 Mainland EN 0.50285714
13 Mainland EF 0.27119377
14 Mainland ZMP 0.09500000
15 Mainland AML 0.53854875
16 Mainland ATPS 0.25038580
17 Mainland MP20 0.80670340
18 Peninsula LTRS 0.42591805
19 Peninsula WNT 0.50613781
20 Peninsula EN 0.17229600
21 Peninsula EF 0.48979592
22 Peninsula ZMP 0.41492187
23 Peninsula AML 0.72842417
24 Peninsula ATPS 0.52255763
25 Peninsula MP20 0.69148800library(ggplot2)
ggplot(he_by_pop, aes(x = Stratum, y = He, fill = Stratum)) +
geom_boxplot() +
theme_minimal() +
labs(title = "Expected Heterozygosity by Species",
y = "He") +
theme(legend.position = "none")